
New open-access platform helps researchers understand genetic changes
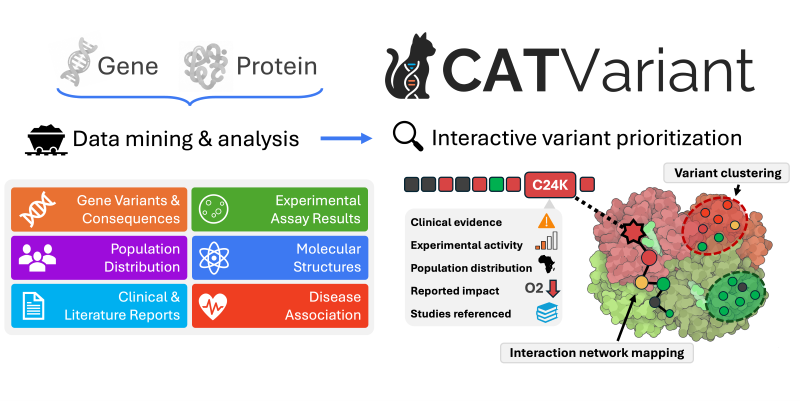
Every person carries small differences in their DNA. Some of these differences, often called variants, do very little. Others can change the amino-acid sequence of a protein, which may affect how that protein is built, folded, moved inside the cell, or how well it does its job. Because proteins carry out many of the body’s essential functions, even a small change can sometimes have important biological or medical consequences.
The challenge is figuring out which changes matter. Researchers often need to weigh many different clues, including whether a variant is rare or common in human populations, whether it has been reported in people with disease, whether it falls in an important or highly conserved part of the protein, whether it may change the protein’s shape or nearby interactions, what experiments have measured, and what the scientific literature says. Each type of evidence has strengths and caveats, and no single source is usually enough on its own. In practice, this often means moving between many separate databases and analysis tools, then manually piecing together a fragmented trail of evidence to decide whether a variant is likely to be harmless, disruptive, or still uncertain.
CATVariant was created to make that process easier and more informative. The platform uses automated data mining to retrieve and organize variant-related evidence from genetic variant databases, protein resources, population datasets, experimental assay collections, disease and pharmacology knowledge bases, and the scientific literature. It then goes further by mapping variants onto the protein sequence and available protein models, comparing them with known functional regions and nearby reported changes, and analyzing broader patterns such as mutation-sensitive regions, structural clusters, and residue connections across the protein. The result is an interactive report that helps users move from a broad protein-level view to detailed review of individual variants without manually stitching the evidence together across multiple resources.
CATVariant is especially useful when direct laboratory or clinical evidence is limited, which is true for many variants. The platform brings together a broad set of computational predictors, with 12 directly surfaced predictor or effect-estimation inputs, and interprets them alongside the rest of the evidence rather than in isolation. These models draw on different kinds of biological signal, including evolutionary conservation, protein sequence patterns, biochemical context, protein shape, and RNA splicing. Because the models capture different signals, CATVariant lets users see where the computational evidence agrees, where it conflicts, and how those predictions line up with structural, population, experimental, and literature evidence.
In short, CATVariant is designed to help researchers turn scattered clues into testable ideas about how a genetic change might affect protein function. The platform is open access and free to use.
NIH Office of Research Infrastructure Programs (ORIP) Workshop

Click on the images to enlarge.


June 2026 — We were honored to participate in the NIH Office of Research Infrastructure Programs (ORIP) Workshop on Advancing Infrastructure to Study Human Diseases, contributing to discussions on the future of innovative models and approaches for biomedical research.
During the In Silico Technologies session, Director Colleen Clancy presented "Digital Twins for the Win: From Molecules to Medicine," highlighting how digital twin technologies integrate mechanistic models, AI, and multimodal data to improve our understanding of disease, accelerate therapeutic discovery, and advance precision medicine.
Thank you to the NIH ORIP organizers and fellow participants for a stimulating conversation about the infrastructure needed to enhance the translatability, reproducibility, and impact of biomedical research.
#DigitalTwins #PrecisionMedicine #ComputationalBiology #SystemsBiology #BiomedicalResearch #NIH #TranslationalScience
Director Colleen Clancy participated in a special symposium at Columbia University
Click on the images to enlarge.


June 2026 — Director Colleen Clancy was honored to participate in a special symposium at Columbia University celebrating the remarkable career and legacy of Professor Robert S. Kass on the occasion of his retirement and 80th birthday.
The symposium brought together former trainees, collaborators, and colleagues to recognize Professor Kass's groundbreaking contributions to cardiovascular science and his extraordinary impact as a mentor. Director Clancy joined fellow speakers in reflecting on the scientific, professional, and personal influence Rocky has had on generations of researchers while sharing advances inspired by his enduring legacy.
Congratulations to Professor Kass on an exceptional career of discovery, leadership, and mentorship. His influence will continue to shape the field and the scientists he has guided for years to come.
Emilie Roncali was a Keynote Speaker at the Virtual Imaging Trials in Medicine Workshop 2026

April 2026 — Associate Director for Computational Biomedicine, Emilie Roncali recently gave a keynote on theranostics digital twins and in silico models at the Virtual Imaging Trials in Medicine workshop 2026. This was a two day online gathering of the Virtual Imaging Trials in Medicine Community for virtual discussions and scientific exchange in the world of in-silico trials, digital twins, and quantitative imaging.
In her talk, Roncali introduced core principles of theranostics digital twins, stressing the difference with in silico models. She discussed examples related to nuclear medicine and detailed some of her research on liver digital twins for liver cancer treatment that integrate computational fluid dynamics and radiation physics modeling to optimize treatment. She briefly discussed some ethical considerations to build diverse, equitable, and accessible digital twins.
Society welcomes inaugural Editors-in-Chief for The Journal of Precision Medicine: Health and Disease and The Journal of Nutritional Physiology

Following the announcement of The Physiological Society’s partnership with Elsevier to launch a new journal, we are delighted to introduce the Editor-in-Chief (Colleen E. Clancy) and Deputy Editor in Chief (Vladimir Yarov-Yarovoy) of The Journal of Precision Medicine: Health and Disease.