
Resident Program - Case of the Month
December 2017 - Presented by Miao Tian (Mentored by Denis Dwyre)
Diagnosis
Acute promyelocytic leukemia (APL) is a subtype of acute myeloid leukemia (AML) in which there is an abnormal accumulation of immature granulocytes called promyelocytes. The FAB classification refers to it as AML M3, whereas the present WHO classification lists it under "Acute myeloid leukemia with recurrent genetic abnormalities." It is characterized by a chromosomal translocation involving the retinoic acid receptor alpha (RARα or RARA) gene on chromosome 17 and the promyelocytic leukemia gene (PML) on chromosome 15. Both hypergranular (so-called typical) APL and microgranular (hypogranular) types exist.
Acute promyelocytic leukemia accounts for 5-8% of AML cases in younger patients, with a lower relative frequency in elderly patients. The disease can occur at any age, but most patients are middle aged adults. The annual incidence rate is 0.08 cases per 100,000 population.
Acute promyelocytic leukemia was first characterized in 1957 by French and Norwegian physicians as a hyperacute fatal illness, with a median survival time of less than a week. It is frequently associated with disseminated intravascular coagulation and increased fibrinolysis. Coagulopathy is associated with significant early death rates in APL patients. Today, prognosis has drastically improved due to the use of all-trans retinoic acid (ATRA); 10-year survival rates are estimated to be approximately 77%. Based upon the initial marrow review, all-trans retinoic acid therapy is often initiated if there is a clinical suspicion for APL. Relapse rates are extremely low.
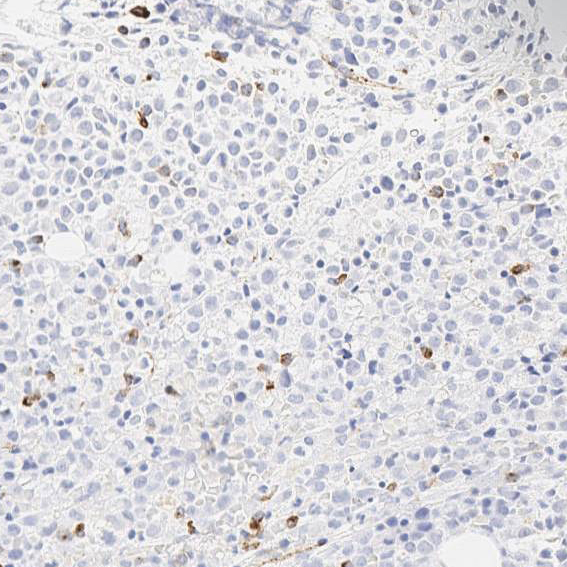
The sensitivity of APL cells to tretinoin (all-trans retinoic acid) has led to the discovery that the RARA gene on 17q21.2 fuses with a nuclear regulatory factor gene on 15q24.1 (PML), giving rise to a PML-RARA fusion gene product. In this case, molecular and cytogenetic studies showed that the patient had a t(15;17)(q24;q21) translocation, confirming the diagnosis of APL. The blasts/blast equivalents in APL with PML-RARA are characterized by low or absent expression of HLA-DR, CD34 and the leukocyte integrins CD11a, CD11b, CD11c and CD18. The granulocytic differentiation markers CD15 and CD65 are negative or only weakly expressed, and CD64 expression is common. In this case, although the flow cytometry study showed heterogeneous CD34 (Figure 4), the CD34 immunostochemical stains on bone marrow biopsy show no CD34 staining on the abnormal promyelocytes (Figure 5).
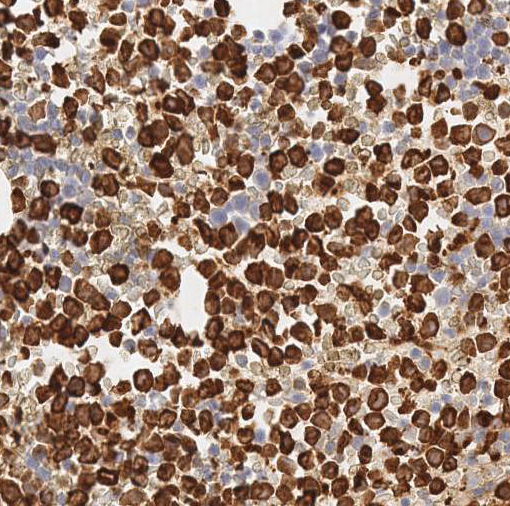
Cytologically, the APL cells are hypergranular promyelocytes with abundant cytoplasm. The nuclei are round or oval with frequently eccentric with moderately condensed chromatin and indistinct nucleoli. Occasional kidney-shaped or bi-lobed nuclei are seen (Figure 2). The cytoplasmic granules may be large and numerous and they may totally obscure the nuclear cytoplasmic margin. Characteristic cells containing bundles of Auer rods randomly distributed within the cytoplasm are present in most cases. Historically, multiple myeloid cytochemical stains (myeloperoxidase, chloracetate esterase and Sudan Black B) have been used in order to highlight the myeloid granules in acute myeloid leukemia, including Auer rods. The MPO reaction is always strongly positive in all the leukemic promyelocytes (Figure 6). In our experience, chloracetate esterase (CAE) cytochemical staining allows the best visualization of the numerous Auer rods typically seen in APL, providing additional data when there is a suspicion for APL. In this case, the collections of Auer rods stained by CAE showed an unusual stellate appearance (Figure 3).
Acute promyelocytic leukemia can be distinguished from other hematological diseases based on microscopic examination of the blood film or a bone marrow aspirate or biopsy as well as finding the characteristic rearrangement. Definitive diagnosis requires the detection of the PML/RARA fusion gene. This may be done by polymerase chain reaction (PCR), fluorescent in situ hybridization (FISH), or conventional cytogenetics of peripheral blood or bone marrow. This mutation typically involves a translocation of the long arm of chromosomes 15 and 17. On rare occasions, a cryptic translocation may occur which cannot be detected by cytogenetic testing; on these occasions PCR testing is essential to confirm the diagnosis. Presence of multiple Auer rods in the malignant cells is highly suggestive of acute promyelocytic leukemia.
Click on image to enlarge.
| Figure 5 | Figure 6 | |
 |
 |
References
- Hillestad, LK (November 1957). "Acute promyelocytic leukemia". Acta Med Scand. 159 (3): 189–94. doi:10.1111/j.0954-6820.1957.tb00124.x. PMID 13508085.
- Tallman MS, Altman JK (2008). "Curative strategies in acute promyelocytic leukemia". Hematology Am Soc Hematol Educ Program. 2008: 391–9.
- Kotiah, SD; Besa, EC (3 June 2013). Sarkodee-Adoo, C; Talavera, F; Sacher, RA; McKenna, R; Besa, EC, eds. "Acute Promyelocytic Leukemia". Medscape Reference. WebMD. Retrieved 14 January 2014
- Coombs, C. C.; Tavakkoli, M.; Tallman, M. S. (2015-04-17). "Acute promyelocytic leukemia: where did we start, where are we now, and the future". Blood Cancer Journal. 5 (4): e304.
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th edition
 Meet our Residency Program Director
Meet our Residency Program Director
